Free energy along drug-protein binding pathways interactively sampled in virtual reality
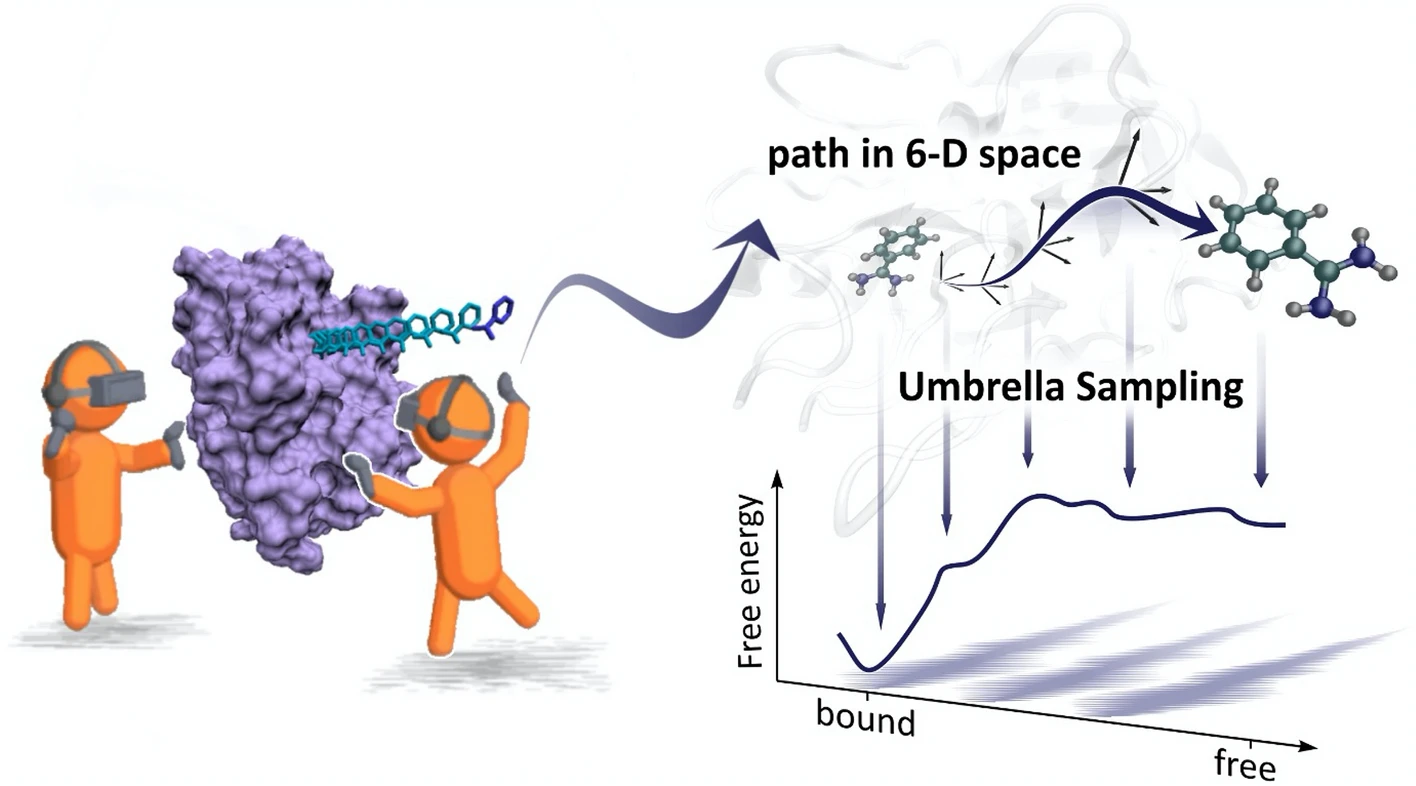
We develop tools to view and interact with molecular simulations in Virtual Reality (VR). These tools let us see molecular systems in a different light to get a new intuition for them. In particular, it gives us a sense of how 3-dimensional and dynamic these systems are. So far, we have mostly gained qualitative insight into the biology and chemistry questions we had about molecular systems. Our new article, in collaboration with the University of Bristol and the University of Valencia, presents a new way to use trajectories generated in Virtual Reality as a base to compute free energies and get quantitative insight.
In this article, published in Scientific Reports, we looked at the trypsin-benzamidine protein-ligand complex. We generated binding and unbinding paths by guiding the ligand in and out of the protein in Virtual Reality. We then computed the free energy along these paths using a 6-dimensional descriptor for the position and orientation of the benzamidine molecule relative to the trypsin. We also used the adaptive string method to optimise the VR-generated paths.
We found that VR-generated paths and free energy calculations can effectively combine to get quantitative insight. We keep looking at ways to improve our Virtual Reality use to better understand molecular systems.